Potenziali classici 1/2
|
Le tecniche di simulazione classica, come la dinamica molecolare, consentono di studiare leproprietà strutturali, statistiche e termodinamiche
di un sistema fisico, e sono basate sul concetto di potenziali interatomici classici.
Questo concetto racchiude un ulteriore e drastico passo di approssimazione
alla complicata equazione che
descrive un sistema di nuclei ed
elettroni: gli elettroni spariscono completamente dalla descrizione, e i loro
effetti vengono modellati da un potenziale di interazione "efficace" tra i
nuclei. Il potenziale sarà dunque una funzione delle sole posizioni
degli atomi, considerati come punti materiali aventi una certa massa.
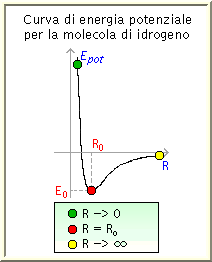
Si tratta di determinare la forma della curva dell'energia
potenziale in funzione delle posizioni degli atomi, considerati come punti
materiali aventi una certa massa.
La curva dell'energia potenziale dipende chiaramente dal tipo di materiale
che stiamo simulando.
I punti sulla curva a determinate distanze interatomiche
aprono una spiegazione sulle caratteristiche del potenziale.
Per i cristalli di gas rari (Argon, Cripton...) si ottengono dei buoni
risultati assumendo che gli atomi interagiscano a coppie (vedi l'esempio in figura).
Però la gran parte dei materiali di interesse pratico (metalli, semiconduttori...)
non può essere descritta in questo modo; in questi casi, l'interazione
fra due atomi dipende anche dalla presenza e dalla posizione degli altri
atomi, cioè bisogna tener conto dell'effetto a molti corpi.
Segui i link ipertestuali della pagina per vedere i dettagli di queste tecniche d'analisi. |